7 Statistics in R

In addition to R’s powerful data manipulation and graphics facilities, R includes a host of procedures which you can use to analyse your data. Many of these procedures are included with the base installation of R, however, even more can be installed with packages available All of the analyses described in this Chapter can be carried out without installing additional packages.
7.1 One and two sample tests
The two main functions for these types of tests are the t.test() and wilcox.test() that perform t tests and Wilcoxon’s signed rank test respectively. Both of these tests can be applied to one and two sample analyses as well as paired data.
As an example of a one sample t test we will use the trees dataset which is included with R. To access this in-built dataset we can use the data() function. This data set provides measurements of the diameter, height and volume of timber in 31 felled black cherry trees (see ?trees for more detail).
# install.packages('skimr') install.packages('gtExtras')
# get the inbult data set of trees
data(trees)
# get the general overview of the data using
str(trees)
## 'data.frame': 31 obs. of 3 variables:
## $ Girth : num 8.3 8.6 8.8 10.5 10.7 10.8 11 11 11.1 11.2 ...
## $ Height: num 70 65 63 72 81 83 66 75 80 75 ...
## $ Volume: num 10.3 10.3 10.2 16.4 18.8 19.7 15.6 18.2 22.6 19.9 ...
# Or
skimr::skim(trees)| Name | trees |
| Number of rows | 31 |
| Number of columns | 3 |
| _______________________ | |
| Column type frequency: | |
| numeric | 3 |
| ________________________ | |
| Group variables | None |
Variable type: numeric
| skim_variable | n_missing | complete_rate | mean | sd | p0 | p25 | p50 | p75 | p100 | hist |
|---|---|---|---|---|---|---|---|---|---|---|
| Girth | 0 | 1 | 13.25 | 3.14 | 8.3 | 11.05 | 12.9 | 15.25 | 20.6 | ▃▇▃▅▁ |
| Height | 0 | 1 | 76.00 | 6.37 | 63.0 | 72.00 | 76.0 | 80.00 | 87.0 | ▃▃▆▇▃ |
| Volume | 0 | 1 | 30.17 | 16.44 | 10.2 | 19.40 | 24.2 | 37.30 | 77.0 | ▇▅▁▂▁ |
# generate a summary of the data using
summary(trees)
## Girth Height Volume
## Min. : 8.30 Min. :63 Min. :10.20
## 1st Qu.:11.05 1st Qu.:72 1st Qu.:19.40
## Median :12.90 Median :76 Median :24.20
## Mean :13.25 Mean :76 Mean :30.17
## 3rd Qu.:15.25 3rd Qu.:80 3rd Qu.:37.30
## Max. :20.60 Max. :87 Max. :77.00
# Or
gtExtras::gt_plt_summary(trees)| trees | ||||||
| 31 rows x 3 cols | ||||||
| Column | Plot Overview | Missing | Mean | Median | SD | |
|---|---|---|---|---|---|---|
| Girth | 0.0% | 13.2 | 12.9 | 3.1 | ||
| Height | 0.0% | 76.0 | 76.0 | 6.4 | ||
| Volume | 0.0% | 30.2 | 24.2 | 16.4 | ||
If we wanted to test whether the mean height of black cherry trees in this sample is equal to 70 ft (mu = 70), assuming these data are normally distributed, we can use a t.test() to do so.
t.test(trees$Height, mu = 70)
##
## One Sample t-test
##
## data: trees$Height
## t = 5.2429, df = 30, p-value = 1.173e-05
## alternative hypothesis: true mean is not equal to 70
## 95 percent confidence interval:
## 73.6628 78.3372
## sample estimates:
## mean of x
## 76The above summary has a fairly logical layout and includes the name of the test that we have asked for (One Sample t-test), which data has been used (data: trees$Height), the t statistic, degrees of freedom and associated p value (t = 5.2, df = 30, p-value = 1e-05). It also states the alternative hypothesis (alternative hypothesis: true mean is not equal to 70) which tells us this is a two sided test (as we have both equal and not equal to), the 95% confidence interval for the mean (95 percent confidence interval:73.66 78.34) and also an estimate of the mean (sample estimates: mean of x : 76). In the above example, the p value is very small and therefore we would reject the null hypothesis and conclude that the mean height of our sample of black cherry trees is not equal to 70 ft.
The function t.test() also has a number of additional arguments which can be used for one-sample tests. You can specify that a one sided test is required by using either alternative = "greater" or alternative = "less arguments which tests whether the sample mean is greater or less than the mean specified. For example, to test whether our sample mean is greater than 70 ft.
t.test(trees$Height, mu = 70, alternative = "greater")
##
## One Sample t-test
##
## data: trees$Height
## t = 5.2429, df = 30, p-value = 5.866e-06
## alternative hypothesis: true mean is greater than 70
## 95 percent confidence interval:
## 74.05764 Inf
## sample estimates:
## mean of x
## 76You can also change the confidence level used for estimating the confidence intervals using the argument conf.level = 0.99. If specified in this way, 99% confidence intervals would be estimated.
Although t tests are fairly robust against small departures from normality you may wish to use a rank based method such as the Wilcoxon’s signed rank test. In R, this is done in almost exactly the same way as the t test but using the wilcox.test() function.
wilcox.test(trees$Height, mu = 70)
##
## Wilcoxon signed rank test with continuity correction
##
## data: trees$Height
## V = 419.5, p-value = 0.0001229
## alternative hypothesis: true location is not equal to 70Don’t worry too much about the warning message, R is just letting you know that your sample contained a number of values which were the same and therefore it was not possible to calculate an exact p value. This is only really a problem with small sample sizes. You can also use the arguments alternative = "greater" and alternative = "less".
In our one sample test it’s always a good idea to examine your data for departures from normality, rather than just assuming everything is OK. Perhaps the simplest way to assess normality is the ‘quantile-quantile plot’. This graph plots the ranked sample quantiles from your distribution against a similar number of ranked quantiles taken from a normal distribution. If your data are normally distributed then the plot of your data points will be in a straight line. Departures from normality will show up as a curve or s-shape in your data points. Judging just how much departure is acceptable comes with a little bit of practice.
To construct a Q-Q plot you need to use both the qqnorm()and qqline()functions. The lty = 2 argument changes the line to a dashed line.
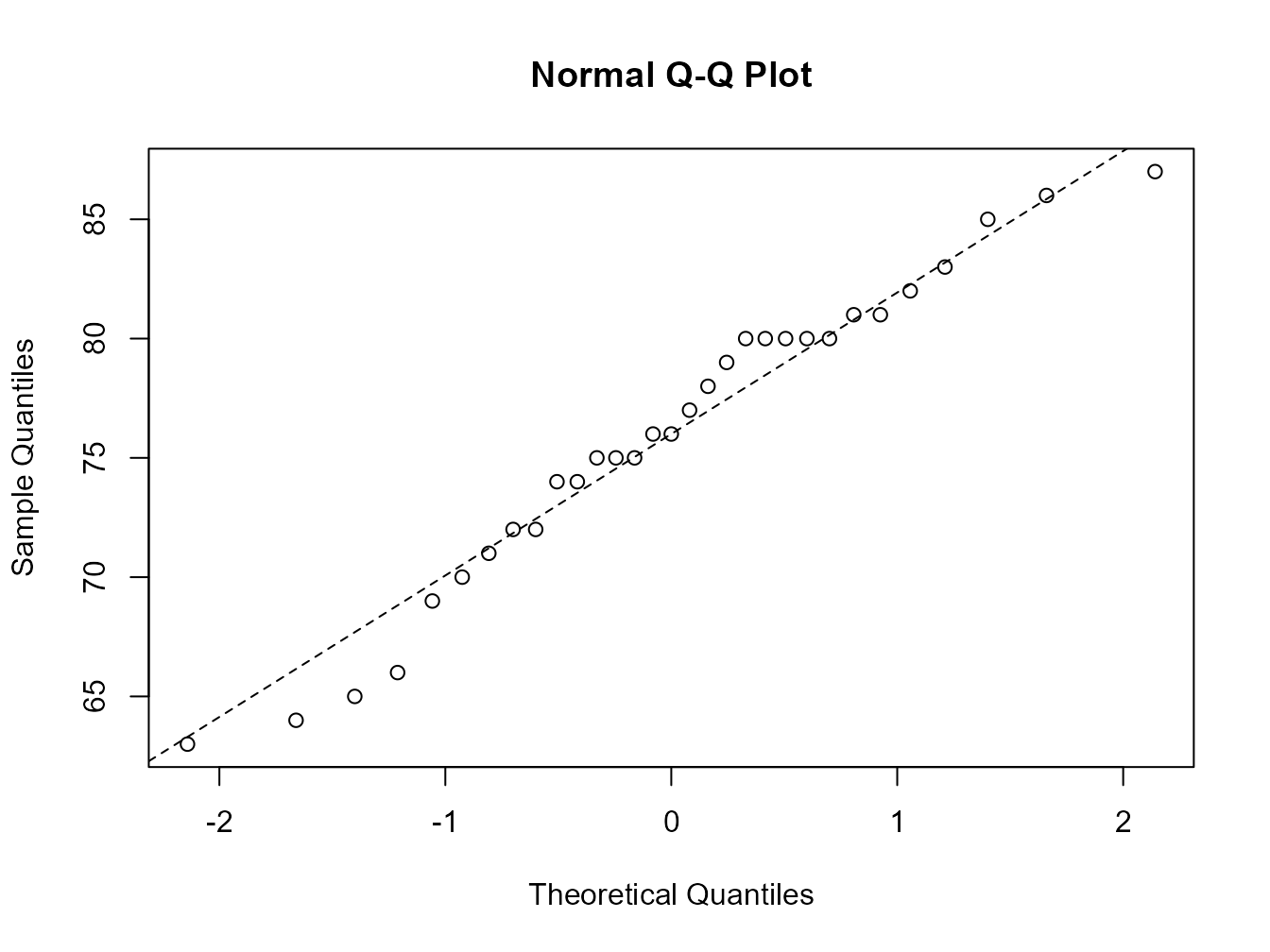
If you insist on performing a specific test for normality you can use the function shapiro.test() which performs a Shapiro – Wilks test of normality.
shapiro.test(trees$Height)
##
## Shapiro-Wilk normality test
##
## data: trees$Height
## W = 0.96545, p-value = 0.4034In the example above, the p value = 0.4 which suggests that there is no evidence to reject the null hypothesis and we can therefore assume these data are normally distributed.
In addition to one-sample tests, both the t.test() and wilcox.test() functions can be used to test for differences between two samples. A two sample t test is used to test the null hypothesis that the two samples come from distributions with the same mean (i.e. the means are not different). For example, a study was conducted to test whether ‘seeding’ clouds with dimethylsulphate alters the moisture content of clouds. Ten random clouds were ‘seeded’ with a further ten ‘unseeded’. The dataset can be found in the atmosphere.txt data file located in the link for the data sets for this guide. click here to open the data folder
atmos <- read.table("data/atmosphere.txt", header = TRUE)
str(atmos)
## 'data.frame': 20 obs. of 2 variables:
## $ moisture : num 301 302 299 316 307 ...
## $ treatment: chr "seeded" "seeded" "seeded" "seeded" ...As with our previous data frame (flowers), these data are in the long format. The column moisture contains the moisture content measured in each cloud and the column treatment identifies whether the cloud was seeded or unseeded. To perform a two-sample t test
t.test(atmos$moisture ~ atmos$treatment)
##
## Welch Two Sample t-test
##
## data: atmos$moisture by atmos$treatment
## t = 2.5404, df = 16.807, p-value = 0.02125
## alternative hypothesis: true difference in means between group seeded and group unseeded is not equal to 0
## 95 percent confidence interval:
## 1.446433 15.693567
## sample estimates:
## mean in group seeded mean in group unseeded
## 303.63 295.06Notice the use of the formula method (atmos$moisture ~ atmos$treatment, which can be read as ‘the moisture described by treatment’) to specify the test. You can also use other methods depending on the format of the data frame. Use ?t.test for further details. The details of the output are similar to the one-sample t test. The Welch’s variant of the t test is used by default and does not assume that the variances of the two samples are equal. If you are sure the variances in the two samples are the same, you can specify this using the var.equal = TRUE argument
t.test(atmos$moisture ~ atmos$treatment, var.equal = TRUE)
##
## Two Sample t-test
##
## data: atmos$moisture by atmos$treatment
## t = 2.5404, df = 18, p-value = 0.02051
## alternative hypothesis: true difference in means between group seeded and group unseeded is not equal to 0
## 95 percent confidence interval:
## 1.482679 15.657321
## sample estimates:
## mean in group seeded mean in group unseeded
## 303.63 295.06To test whether the assumption of equal variances is valid you can perform an F-test on the ratio of the group variances using the var.test() function.
var.test(atmos$moisture ~ atmos$treatment)
##
## F test to compare two variances
##
## data: atmos$moisture by atmos$treatment
## F = 0.57919, num df = 9, denom df = 9, p-value =
## 0.4283
## alternative hypothesis: true ratio of variances is not equal to 1
## 95 percent confidence interval:
## 0.1438623 2.3318107
## sample estimates:
## ratio of variances
## 0.5791888As the p value is greater than 0.05, there is no evidence to suggest that the variances are unequal. Note however, that the F-test is sensitive to departures from normality and should not be used with data which is not normal. See the car package for alternatives.
The non-parametric two-sample Wilcoxon test (also known as a Mann-Whitney U test) can be performed using the same formula method:
wilcox.test(atmos$moisture ~ atmos$treatment)
##
## Wilcoxon rank sum exact test
##
## data: atmos$moisture by atmos$treatment
## W = 79, p-value = 0.02881
## alternative hypothesis: true location shift is not equal to 0You can also use the t.test() and wilcox.test() functions to test paired data. Paired data are where there are two measurements on the same experimental unit (individual, site etc) and essentially tests for whether the mean difference between the paired observations is equal to zero. For example, the pollution dataset gives the biodiversity score of aquatic invertebrates collected using kick samples in 17 different rivers. These data are paired because two samples were taken on each river, one upstream of a paper mill and one downstream.
pollution <- read.table("data/pollution.txt", header = TRUE)
str(pollution)
## 'data.frame': 16 obs. of 2 variables:
## $ down: int 20 15 10 5 20 15 10 5 20 15 ...
## $ up : int 23 16 10 4 22 15 12 7 21 16 ...Note, in this case these data are in the wide format with upstream and downstream values in separate columns (see previous topics on how to convert to long format if you want). To conduct a paired t test use the paired = TRUE argument.
t.test(pollution$down, pollution$up, paired = TRUE)
##
## Paired t-test
##
## data: pollution$down and pollution$up
## t = -3.0502, df = 15, p-value = 0.0081
## alternative hypothesis: true mean difference is not equal to 0
## 95 percent confidence interval:
## -1.4864388 -0.2635612
## sample estimates:
## mean difference
## -0.875The output is almost identical to that of a one-sample t test. It is also possible to perform a non-parametric matched-pairs Wilcoxon test in the same way
wilcox.test(pollution$down, pollution$up, paired = TRUE)
##
## Wilcoxon signed rank test with continuity correction
##
## data: pollution$down and pollution$up
## V = 8, p-value = 0.01406
## alternative hypothesis: true location shift is not equal to 0The function prop.test() can be used to compare two or more proportions. For example, a company wishes to test the effectiveness of an advertising campaign for a particular brand of cat food. The company commissions two polls, one before the advertising campaign and one after, with each poll asking cat owners whether they would buy this brand of cat food. The results are given in the table below
| before | after | |
|---|---|---|
| would buy | 45 | 71 |
| would not buy | 35 | 32 |
From the table above we can conclude that 56% of cat owners would buy the cat food before the campaign compared to 69% after. But, has the advertising campaign been a success?
The prop.test() function has two main arguments which are given as two vectors. The first vector contains the number of positive outcomes and the second vector the total numbers for each group. So to perform the test we first need to define these vectors
buy <- c(45, 71) # creates a vector of positive outcomes
total <- c((45 + 35), (71 + 32)) # creates a vector of total numbers
prop.test(buy, total) # perform the test
##
## 2-sample test for equality of proportions with
## continuity correction
##
## data: buy out of total
## X-squared = 2.598, df = 1, p-value = 0.107
## alternative hypothesis: two.sided
## 95 percent confidence interval:
## -0.27865200 0.02501122
## sample estimates:
## prop 1 prop 2
## 0.5625000 0.6893204There is no evidence to support that the advertising campaign has changed cat owners opinions of the cat food (p = 0.1). Use ?prop.test to explore additional uses of the binomial proportions test.
We could also analyse the count data in the above example as a Chi-square contingency table. The simplest method is to convert the tabulated table into a 2 x 2 matrix using the matrix() function (note, there are many alternative methods of constructing a table like this).
Notice that you enter the data column wise into the matrix and then specify the number of rows using nrow =.
We can also change the row names and column names from the defaults to make it look more like a table (you don’t really need to do this to perform a Chi-square test)
colnames(buyers) <- c("before", "after")
rownames(buyers) <- c("buy", "notbuy")
buyers
## before after
## buy 45 71
## notbuy 35 32You can then perform a Chi-square test to test whether the number of cat owners buying the cat food is independent of the advertising campaign using the chisq.test() function. In this example the only argument is our matrix of counts.
chisq.test(buyers)
##
## Pearson's Chi-squared test with Yates' continuity
## correction
##
## data: buyers
## X-squared = 2.598, df = 1, p-value = 0.107There is no evidence (p = 0.107) to suggest that we should reject the null hypothesis that the number of cat owners buying the cat food is independent of the advertising campaign. You may have spotted that for a 2x2 table, this test is exactly equivalent to the prop.test(). You can also use the chisq.test() function on raw (untabulated) data and to test for goodness of fit (see ?chisq.test for more details).
7.2 Correlation
In R, the Pearson’s product-moment correlation coefficient between two continuous variables can be estimated using the cor() function. Using the trees data set again, we can determine the correlation coefficient of the association between tree Height and Volume.
data(trees)
str(trees)
## 'data.frame': 31 obs. of 3 variables:
## $ Girth : num 8.3 8.6 8.8 10.5 10.7 10.8 11 11 11.1 11.2 ...
## $ Height: num 70 65 63 72 81 83 66 75 80 75 ...
## $ Volume: num 10.3 10.3 10.2 16.4 18.8 19.7 15.6 18.2 22.6 19.9 ...
cor(trees$Height, trees$Volume)
## [1] 0.5982497or we can produce a matrix of correlation coefficients for all variables in a data frame
cor(trees)
## Girth Height Volume
## Girth 1.0000000 0.5192801 0.9671194
## Height 0.5192801 1.0000000 0.5982497
## Volume 0.9671194 0.5982497 1.0000000Note that the correlation coefficients are identical in each half of the matrix. Also, be aware that, although a matrix of coefficients can be useful, a little commonsense should be used when using cor() on data frames with numerous variables. It is not good practice to trawl through these types of matrices in the hope of finding large coefficients without having an a priori reason for doing so and remember the correlation coefficient assumes that associations are linear.
If you have missing values in the variables you are trying to correlate, cor() will return an error message (as will many functions in R). You will either have to remove these observations (be very careful if you do this) or tell R what to do when an observation is missing. A useful argument you can use with the cor() function is use = "complete.obs".
cor(trees, use = "complete.obs")
## Girth Height Volume
## Girth 1.0000000 0.5192801 0.9671194
## Height 0.5192801 1.0000000 0.5982497
## Volume 0.9671194 0.5982497 1.0000000The function cor() will return the correlation coefficient of two variables, but gives no indication whether the coefficient is significantly different from zero. To do this you need to use the function cor.test().
cor.test(trees$Height, trees$Volume)
##
## Pearson's product-moment correlation
##
## data: trees$Height and trees$Volume
## t = 4.0205, df = 29, p-value = 0.0003784
## alternative hypothesis: true correlation is not equal to 0
## 95 percent confidence interval:
## 0.3095235 0.7859756
## sample estimates:
## cor
## 0.5982497Two non-parametric equivalents to Pearson correlation are available within the cor.test() function; Spearman’s rank and Kendall’s tau coefficient. To use either of these simply include the argument method = "spearman" or method = "kendall" depending on the test you wish to use. For example
cor.test(trees$Height, trees$Volume, method = "spearman")
##
## Spearman's rank correlation rho
##
## data: trees$Height and trees$Volume
## S = 2089.6, p-value = 0.0006484
## alternative hypothesis: true rho is not equal to 0
## sample estimates:
## rho
## 0.57871017.3 Simple linear modelling
Linear models are one of the most widely used models in statistics and data science. They are often thought of as simple models but they’re very flexible and able to model a wide variety of experimental and survey designs. Many of the statistical approaches you may have used previously (such as linear regression, t-test, ANOVA, ANCOVA etc) can be expressed as a linear model so the good news is that you’re probably already familiar with linear models (albeit indirectly). They also form the foundation of more complicated modelling approaches and are relatively easy to extended to incorporate additional complexity. During this section we’ll learn how to fit some simple linear models using R and cover some of the more common applications. We won’t go into any detail of the underlying linear modelling theory but rather focus on the practicalities of model fitting and R code.
The main function for fitting linear models in R is the lm() function (short for linear model!). The lm() function has many arguments but the most important is the first argument which specifies the model you want to fit using a model formula which typically takes the general form:
response variable ~ explanatory variable(s)
This model formula is simply read as
‘variation in the response variable modelled as a function (~) of the explanatory variable(s)’.
The response variable is also commonly known as the ‘dependent variable’ and the explanatory variables are sometimes referred to as ‘independent variables’ (or less frequently as ‘predictor variables’). There is also an additional term in our model formula which represents the variation in our response variable not explained by our explanatory variables but you don’t need to specify this when using the lm() function.
As mentioned above, many of the statistical ‘tests’ you might have previously used can be expressed as a linear model. For example, if we wanted to perform a bivariate linear regression between a response variable (y) and a single continuous explanatory variable (x) our model formula would simply be
y ~ xOn the other hand, if we wanted to use an ANOVA to test whether the group means of a response variable (y) were different between a three level factor (x) our model formula would look like
y ~ xOK, hang on, they both look identical, what gives? In addition to the model formula, the type of linear model you fit is also determined by the type of data in your explanatory variable(s) (i.e. what class of data). If your explanatory variable is continuous then you will fit a bivariate linear regression. If your explanatory variable is a factor (i.e. categorical data) you will fit an ANOVA type model.
You can also increase the complexity of your linear model by including additional explanatory variables in your model formula. For example, if we wanted to fit a two-way ANOVA both of our explanatory variables x and z would need to be factors and separated by a + symbol
y ~ x + zIf we wanted to perform a factorial ANOVA to identify an interaction between both explanatory variables we would separate our explanatory variables with a : symbol whilst also including our main effects in our model formula
y ~ x + z + x:zor by using the equivalent shortcut notation
y ~ x * z It’s important that you get comfortable with using model formula (and we’ve only given the briefest of explanations above) when using the lm() function (and other functions) as it’s remarkably easy to specifiy a model which is either nonsense or isn’t the model you really wanted to fit. A summary table of various linear model formula and equivalent R code given below.
| Traditional name | Model formula | R code |
|---|---|---|
| Bivariate regression | Y ~ X1 (continuous) | lm(Y ~ X) |
| One-way ANOVA | Y ~ X1 (categorical) | lm(Y ~ X) |
| Two-way ANOVA | Y ~ X1 (cat) + X2(cat) | lm(Y ~ X1 + X2) |
| ANCOVA | Y ~ X1 (cat) + X2(cont) | lm(Y ~ X1 + X2) |
| Multiple regression | Y ~ X1 (cont) + X2(cont) | lm(Y ~ X1 + X2) |
| Factorial ANOVA | Y ~ X1 (cat) * X2(cat) |
|
OK, time for an example. The data file smoking.txt summarises the results of a study investigating the possible relationship between mortality rate and smoking across 25 occupational groups in the UK. The variable occupational.group specifies the different occupational groups studied, the risk.group variable indicates the relative risk to lung disease for the various occupational groups and smoking is an index of the average number of cigarettes smoked each day (relative to the number smoked across all occupations). The variable mortality is an index of the death rate from lung cancer in each group (relative to the death rate across all occupational groups). In this data set, the response variable is mortality and the potential explanatory variables are smoking which is numeric and risk.group which is a three level factor. The first thing to do is import our data file using the read.table() function as usual and assign the data to an object called smoke. You can find a link to download these data, click here to open the data folder
smoke <- read.table("data/smoking.txt", header = TRUE, sep = "\t",
stringsAsFactors = TRUE)
str(smoke, vec.len = 2)
## 'data.frame': 25 obs. of 4 variables:
## $ occupational.group: Factor w/ 25 levels "Administrators",..: 9 14 2 11 10 ...
## $ risk.group : Factor w/ 3 levels "high","low","medium": 2 1 1 3 1 ...
## $ smoking : int 77 137 117 94 116 ...
## $ mortality : int 84 116 123 128 155 ...
# vec.len argument to limited number of ' first elements'
# to displayNext, let’s investigate the relationship between the mortality and smoking variables by plotting a scatter plot. We can use either the ggplot2 package or base R graphics to do this. We’ll use ggplot2 this time and our old friend the ggplot() function.
library(ggplot2)
ggplot(mapping = aes(x = smoking, y = mortality), data = smoke) +
geom_point()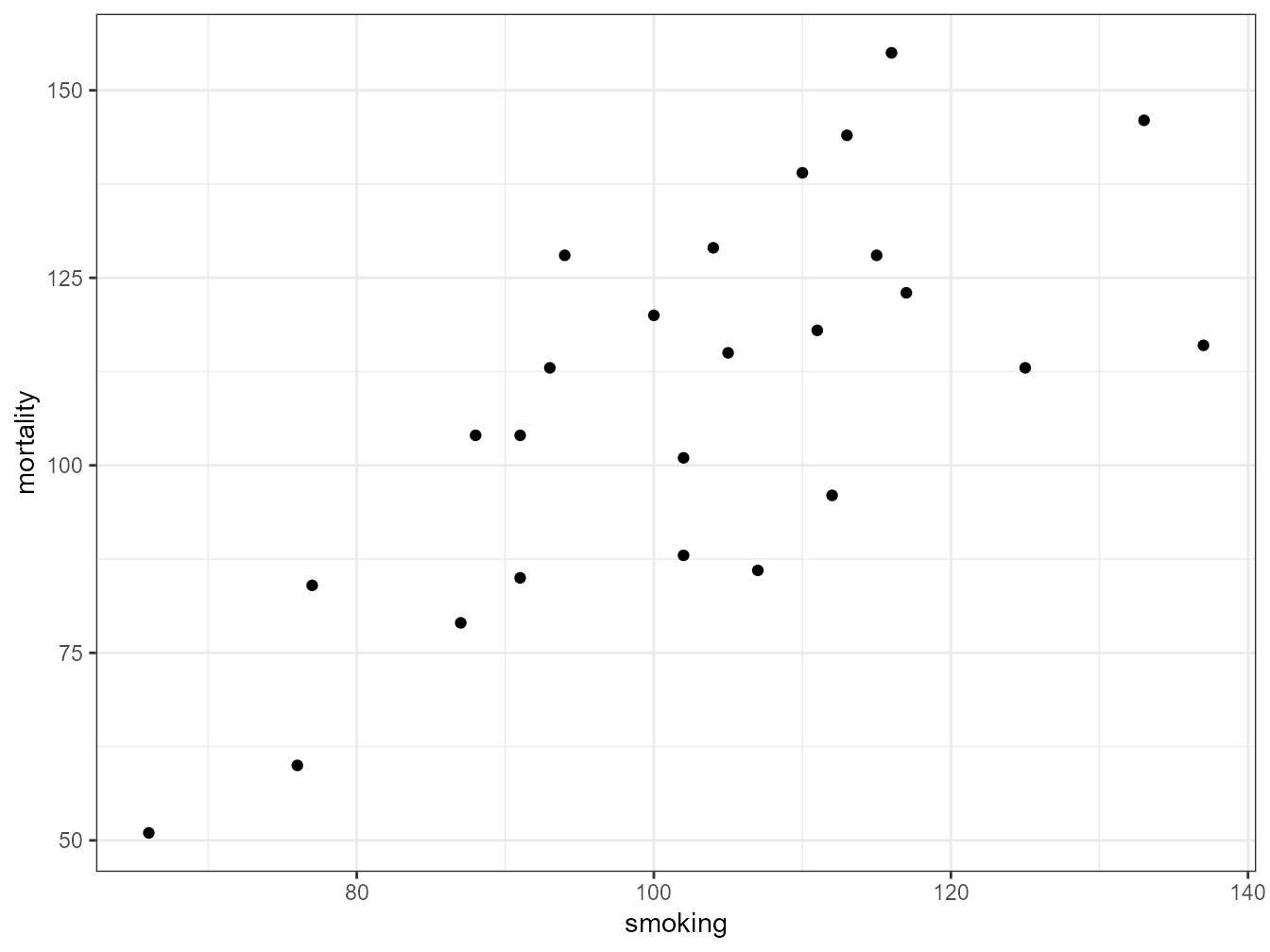
The plot does suggest that there is a positive relationship between the smoking index and mortality index.
To fit a simple linear model to these data we will use the lm() function and include our model formula mortality ~ smoking and assign the results to an object called smoke_lm.
smoke_lm <- lm(mortality ~ smoking, data = smoke)Notice that we have not used the $ notation to specify the variables in our model formula, instead we’ve used the data = smoke argument. Although the $ notation will work (i.e. smoke$mortality ~ smoke$smoking) it will more than likely cause you problems later on and should be avoided. In fact, we would go as far to suggest that if any function has a data = argument you should always use it. How do you know if a function has a data = argument? Just look in the associated help file.
Perhaps somewhat confusingly (at least at first) it appears that nothing much has happened, you don’t automatically get the voluminous output that you normally get with other statistical packages. In fact, what R does, is store the output of the analysis in what is known as a lm class object (which we have called smoke_lm) from which you are able to extract exactly what you want using other functions. If you’re brave, you can examine the structure of the smoke_lm model object using the str() function.
str(smoke_lm)
## List of 12
## $ coefficients : Named num [1:2] -2.89 1.09
## ..- attr(*, "names")= chr [1:2] "(Intercept)" "smoking"
## $ residuals : Named num [1:25] 3.15 -30.11 -1.36 28.66 31.73 ...
## ..- attr(*, "names")= chr [1:25] "1" "2" "3" "4" ...
## $ effects : Named num [1:25] -545 -91.63 2.72 26.99 35.56 ...
## ..- attr(*, "names")= chr [1:25] "(Intercept)" "smoking" "" "" ...
## $ rank : int 2
## $ fitted.values: Named num [1:25] 80.9 146.1 124.4 99.3 123.3 ...
## ..- attr(*, "names")= chr [1:25] "1" "2" "3" "4" ...
## $ assign : int [1:2] 0 1
## $ qr :List of 5
## ..$ qr : num [1:25, 1:2] -5 0.2 0.2 0.2 0.2 0.2 0.2 0.2 0.2 0.2 ...
## .. ..- attr(*, "dimnames")=List of 2
## .. .. ..$ : chr [1:25] "1" "2" "3" "4" ...
## .. .. ..$ : chr [1:2] "(Intercept)" "smoking"
## .. ..- attr(*, "assign")= int [1:2] 0 1
## ..$ qraux: num [1:2] 1.2 1.46
## ..$ pivot: int [1:2] 1 2
## ..$ tol : num 1e-07
## ..$ rank : int 2
## ..- attr(*, "class")= chr "qr"
## $ df.residual : int 23
## $ xlevels : Named list()
## $ call : language lm(formula = mortality ~ smoking, data = smoke)
## $ terms :Classes 'terms', 'formula' language mortality ~ smoking
## .. ..- attr(*, "variables")= language list(mortality, smoking)
## .. ..- attr(*, "factors")= int [1:2, 1] 0 1
## .. .. ..- attr(*, "dimnames")=List of 2
## .. .. .. ..$ : chr [1:2] "mortality" "smoking"
## .. .. .. ..$ : chr "smoking"
## .. ..- attr(*, "term.labels")= chr "smoking"
## .. ..- attr(*, "order")= int 1
## .. ..- attr(*, "intercept")= int 1
## .. ..- attr(*, "response")= int 1
## .. ..- attr(*, ".Environment")=<environment: R_GlobalEnv>
## .. ..- attr(*, "predvars")= language list(mortality, smoking)
## .. ..- attr(*, "dataClasses")= Named chr [1:2] "numeric" "numeric"
## .. .. ..- attr(*, "names")= chr [1:2] "mortality" "smoking"
## $ model :'data.frame': 25 obs. of 2 variables:
## ..$ mortality: int [1:25] 84 116 123 128 155 101 118 113 104 88 ...
## ..$ smoking : int [1:25] 77 137 117 94 116 102 111 93 88 102 ...
## ..- attr(*, "terms")=Classes 'terms', 'formula' language mortality ~ smoking
## .. .. ..- attr(*, "variables")= language list(mortality, smoking)
## .. .. ..- attr(*, "factors")= int [1:2, 1] 0 1
## .. .. .. ..- attr(*, "dimnames")=List of 2
## .. .. .. .. ..$ : chr [1:2] "mortality" "smoking"
## .. .. .. .. ..$ : chr "smoking"
## .. .. ..- attr(*, "term.labels")= chr "smoking"
## .. .. ..- attr(*, "order")= int 1
## .. .. ..- attr(*, "intercept")= int 1
## .. .. ..- attr(*, "response")= int 1
## .. .. ..- attr(*, ".Environment")=<environment: R_GlobalEnv>
## .. .. ..- attr(*, "predvars")= language list(mortality, smoking)
## .. .. ..- attr(*, "dataClasses")= Named chr [1:2] "numeric" "numeric"
## .. .. .. ..- attr(*, "names")= chr [1:2] "mortality" "smoking"
## - attr(*, "class")= chr "lm"To obtain a summary of our analysis we can use the summary() function on our smoke_lm model object.
summary(smoke_lm)
##
## Call:
## lm(formula = mortality ~ smoking, data = smoke)
##
## Residuals:
## Min 1Q Median 3Q Max
## -30.107 -17.892 3.145 14.132 31.732
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) -2.8853 23.0337 -0.125 0.901
## smoking 1.0875 0.2209 4.922 5.66e-05 ***
## ---
## Signif. codes:
## 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 18.62 on 23 degrees of freedom
## Multiple R-squared: 0.513, Adjusted R-squared: 0.4918
## F-statistic: 24.23 on 1 and 23 DF, p-value: 5.658e-05This shows you everything you need to know about the parameter estimates (intercept and slope), their standard errors and associated t statistics and p values. The estimate for the Intercept suggests that when the relative smoking index is 0 the relative mortality rate is -2.885! The p value associated with the intercept tests the null hypothesis that the intercept is equal to zero. As the p value is large we fail to reject this null hypothesis. The smoking parameter estimate (1.0875) is the estimate of the slope and suggests that for every unit increase in the average number of cigarettes smoked each day the mortality risk index increases by 1.0875. The p value associated with the smoking parameter tests whether the slope of this relationship is equal to zero (i.e. no relationship). As our p value is small we reject this null hypothesis and therefore the slope is different from zero and therefore there is a significant relationship. The summary table also includes other important information such as the coefficient of determination (R2), adjusted R2 , F statistic, associated degrees of freedom and p value. This information is a condensed form of an ANOVA table which you can see by using the anova() function.
anova(smoke_lm)
## Analysis of Variance Table
##
## Response: mortality
## Df Sum Sq Mean Sq F value Pr(>F)
## smoking 1 8395.7 8395.7 24.228 5.658e-05 ***
## Residuals 23 7970.3 346.5
## ---
## Signif. codes:
## 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Now let’s fit another linear model, but this time we will use the risk.group variable as our explanatory variable. Remember the risk.group variable is a factor and so our linear model will be equivalent to an ANOVA type analysis. We will be testing the null hypothesis that there is no difference in the mean mortality rate between the low, medium and high groups. We fit the model in exactly the same way as before.
smoke_risk_lm <- lm(mortality ~ risk.group, data = smoke)Again, we can produce an ANOVA table using the anova() function
anova(smoke_risk_lm)
## Analysis of Variance Table
##
## Response: mortality
## Df Sum Sq Mean Sq F value Pr(>F)
## risk.group 2 11514.4 5757.2 26.107 1.554e-06 ***
## Residuals 22 4851.6 220.5
## ---
## Signif. codes:
## 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1The results presented in the ANOVA table suggest that we can reject the null hypothesis (very small p value) and therefore the mean mortality rate index is different between low, medium and high risk groups.
As we did with our first linear model we can also produce a summary of the estimated parameters using the summary() function.
summary(smoke_risk_lm)
##
## Call:
## lm(formula = mortality ~ risk.group, data = smoke)
##
## Residuals:
## Min 1Q Median 3Q Max
## -26.17 -11.45 4.00 9.00 26.83
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 135.00 5.25 25.713 < 2e-16 ***
## risk.grouplow -57.83 8.02 -7.211 3.16e-07 ***
## risk.groupmedium -27.55 6.90 -3.992 0.000615 ***
## ---
## Signif. codes:
## 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 14.85 on 22 degrees of freedom
## Multiple R-squared: 0.7036, Adjusted R-squared: 0.6766
## F-statistic: 26.11 on 2 and 22 DF, p-value: 1.554e-06In the summary table the Intercept is set to the first level of risk.group (high) as this occurs first alphabetically. Therefore, the estimated mean mortality index for high risk individuals is 135. The estimates for risk.grouplow and risk.groupmedium are mean differences from the intercept (high group). So the mortality index for the low group is 135 - 57.83 = 77.17 and for the medium group is 135 - 27.55 = 107.45. The t values and p values in the summary table are associated with testing specific hypotheses. The p value associated with the intercept tests the null hypothesis that the mean mortality index for the high group is equal to zero. To be honest this is not a particularly meaningful hypothesis to test but we can reject it anyway as we have a very small p value. The p value for the risk.grouplow parameter tests the null hypothesis that the mean difference between high and low risk groups is equal to zero (i.e. there is no difference). Again we reject this null hypothesis and conclude that the means are different between these two groups. Similarly, the p value for risk.groupmedium tests the null hypothesis that the mean difference between high and medium groups is equal to zero which we also reject.
Don’t worry too much if you find the output from the summary() function a little confusing. Its takes a bit of practice and experience to be able to make sense of all the numbers. Remember though, the more complicated your model is, the more complicated your interpretion will be. And always remember, a model that you can’t interpret is not worth fitting (most of the time!).
Another approach to interpreting your model output is to plot a graph of your data and then add the fitted model to this plot. Let’s go back to the first linear model we fitted (smoke_lm). We can add the fitted line to our previous plot using the ggplot2 package and the geom_smooth geom. We can easily include the standard errors by specifying the se = TRUE argument.
ggplot(mapping = aes(x = smoking, y = mortality), data = smoke) +
geom_point() + geom_smooth(method = "lm", se = TRUE)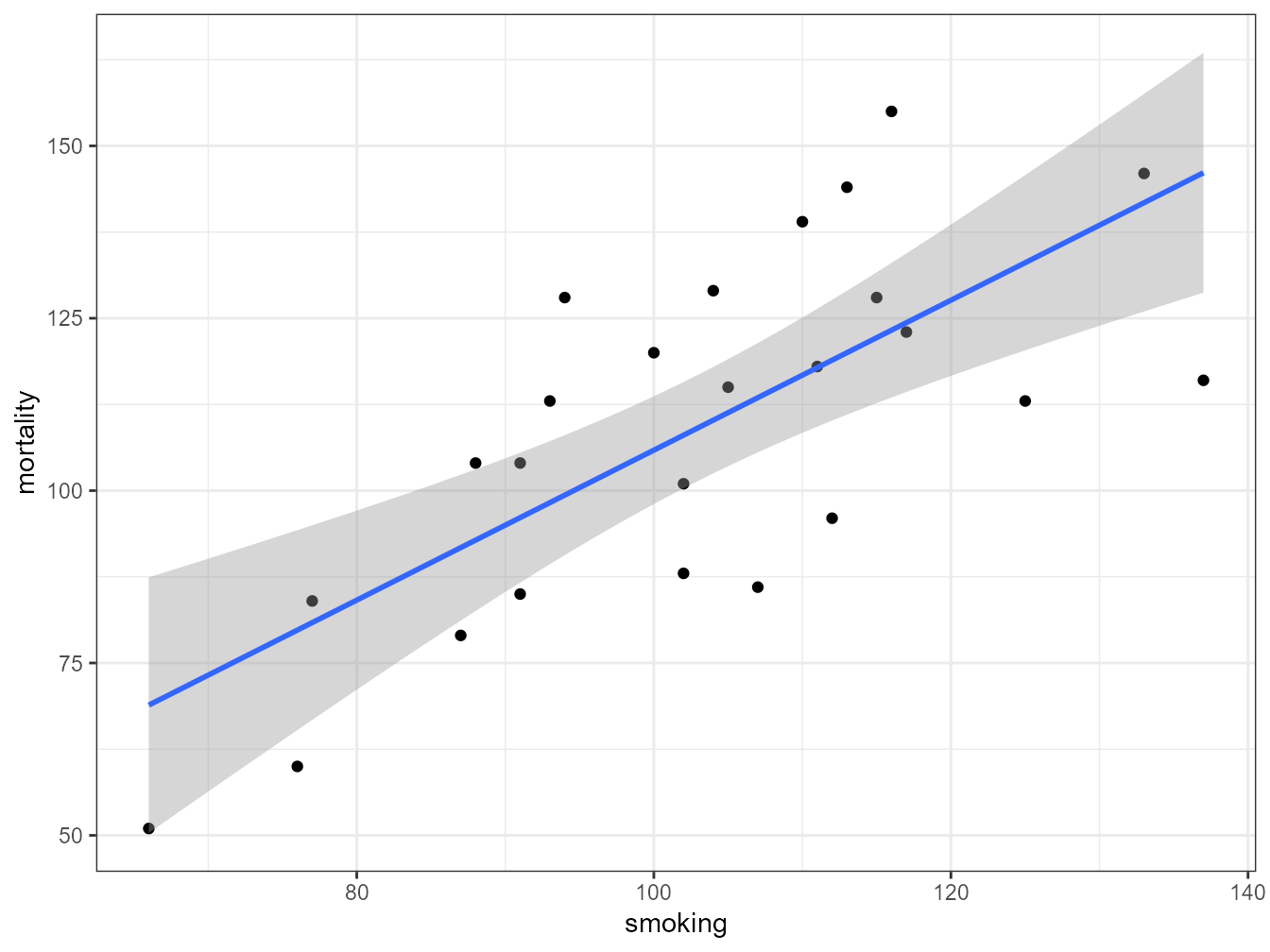
You can also do this with R’s base graphics. Note though that the fitted line extends beyond the data which is not great practice. If you want to prevent this you can generate predicted values from the model using the predict() function within the range of your data and then add these values to the plot using the lines() function (not shown).
plot(smoke$smoking, smoke$mortality, xlab = "smoking rate", ylab = " mortality rate")
abline(smoke_lm, col = "red")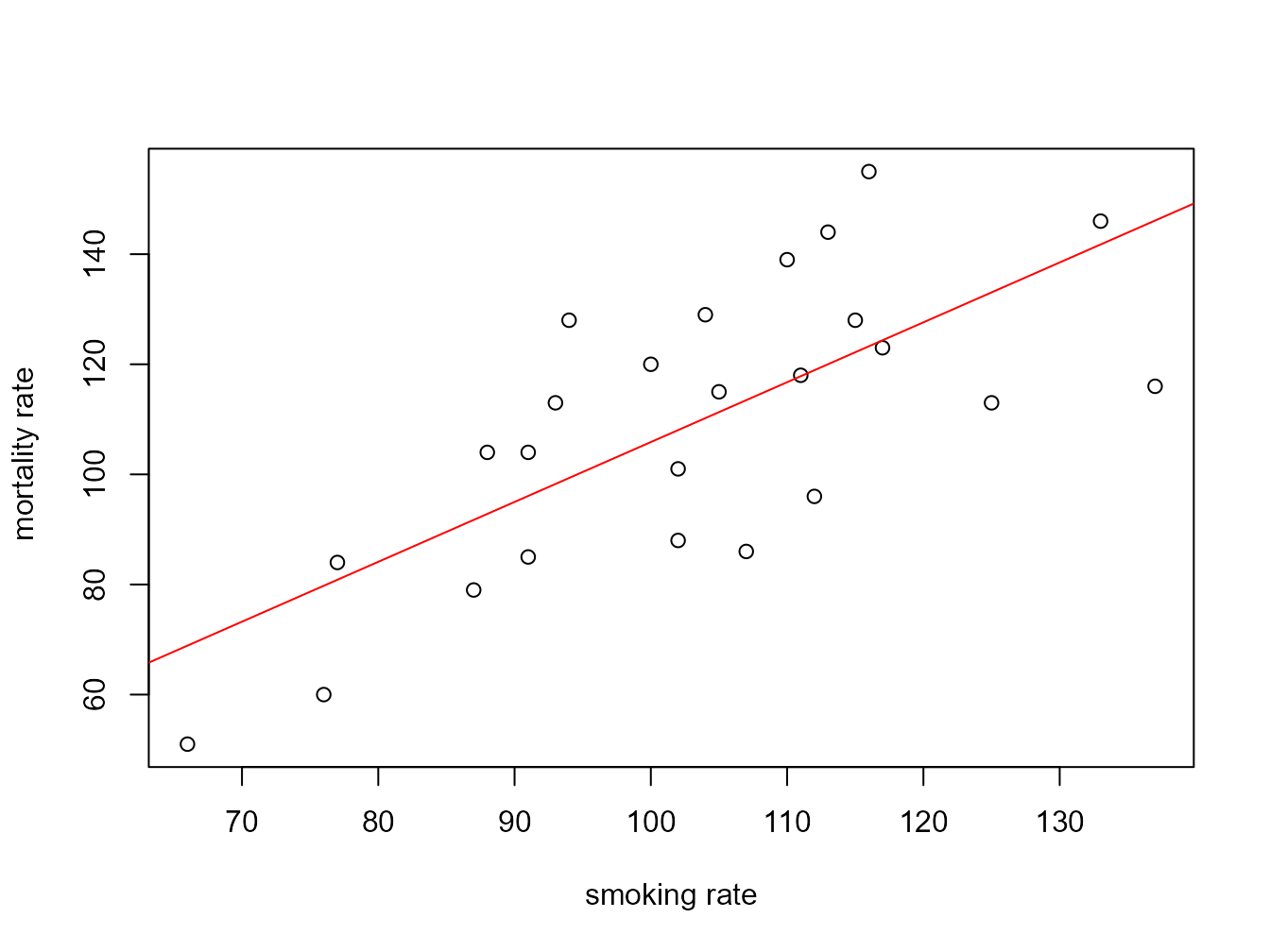
Before we sit back and relax and admire our model (or go write that high impact paper your supervisor/boss has been harassing you about) our work is not finished. It’s vitally important to check the underlying assumptions of your linear model. Two of the most important assumption are equal variances (homogeneity of variance) and normality of residuals. To check for equal variances we can construct a graph of residuals versus fitted values. We can do this by first extracting the residuals and fitted values from our model object using the resid() and fitted() functions.
And then plot them using ggplot or base R graphics.
ggplot(mapping = aes(x = smoke_fit, y = smoke_res)) + geom_point() +
geom_hline(yintercept = 0, colour = "red", linetype = "dashed")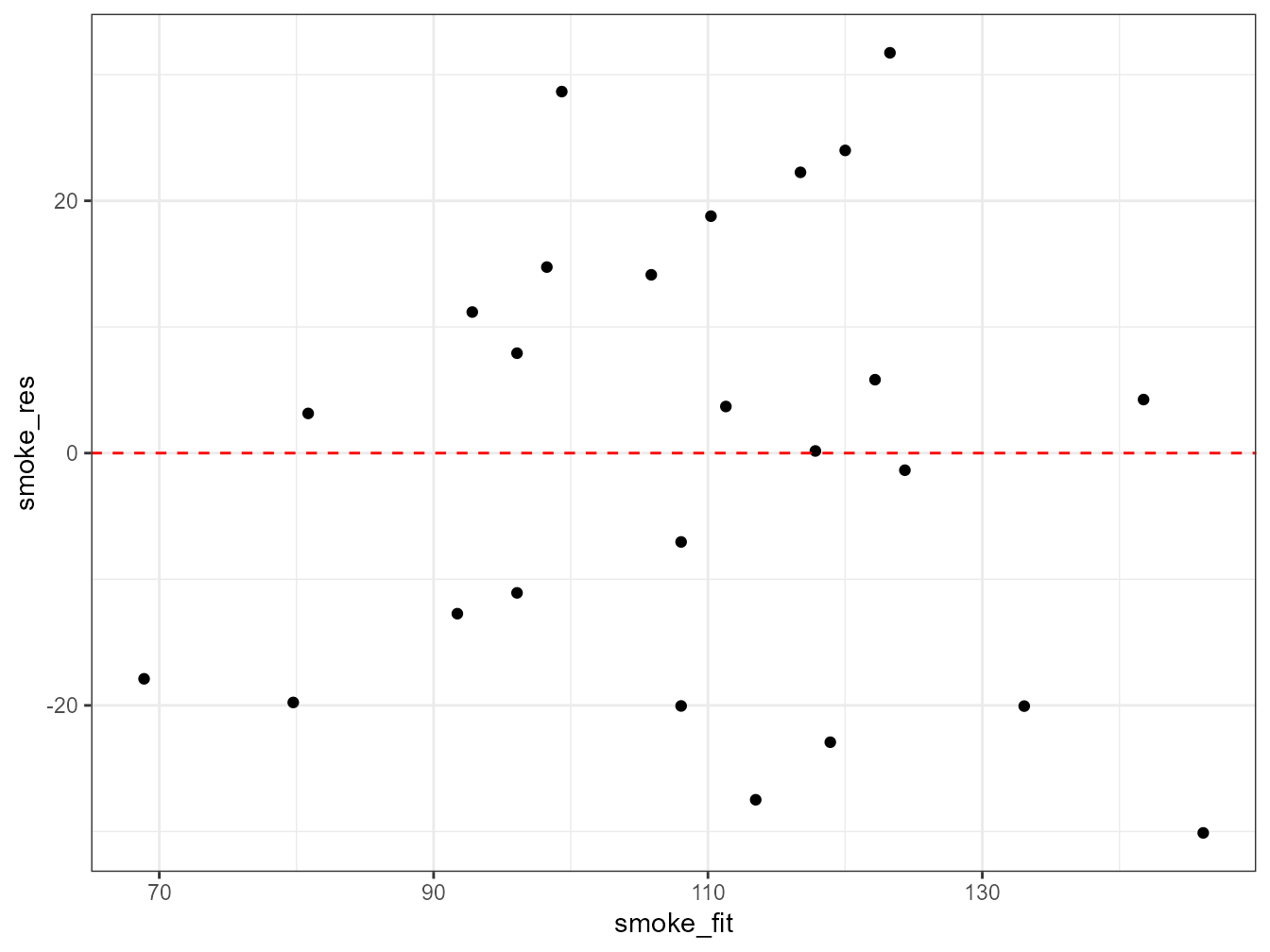
It takes a little practice to interpret these types of graph, but what you are looking for is no pattern or structure in your residuals. What you definitely don’t want to see is the scatter increasing around the zero line (red dashed line) as the fitted values get bigger (this has been described as looking like a trumpet, a wedge of cheese or even a slice of pizza) which would indicate unequal variances (heteroscedacity).
To check for normality of residuals we can use our old friend the Q-Q plot using the residuals stored in the smoke_res object we created earlier.
ggplot(mapping = aes(sample = smoke_res)) + stat_qq() + stat_qq_line()
Or the same plot with base graphics.
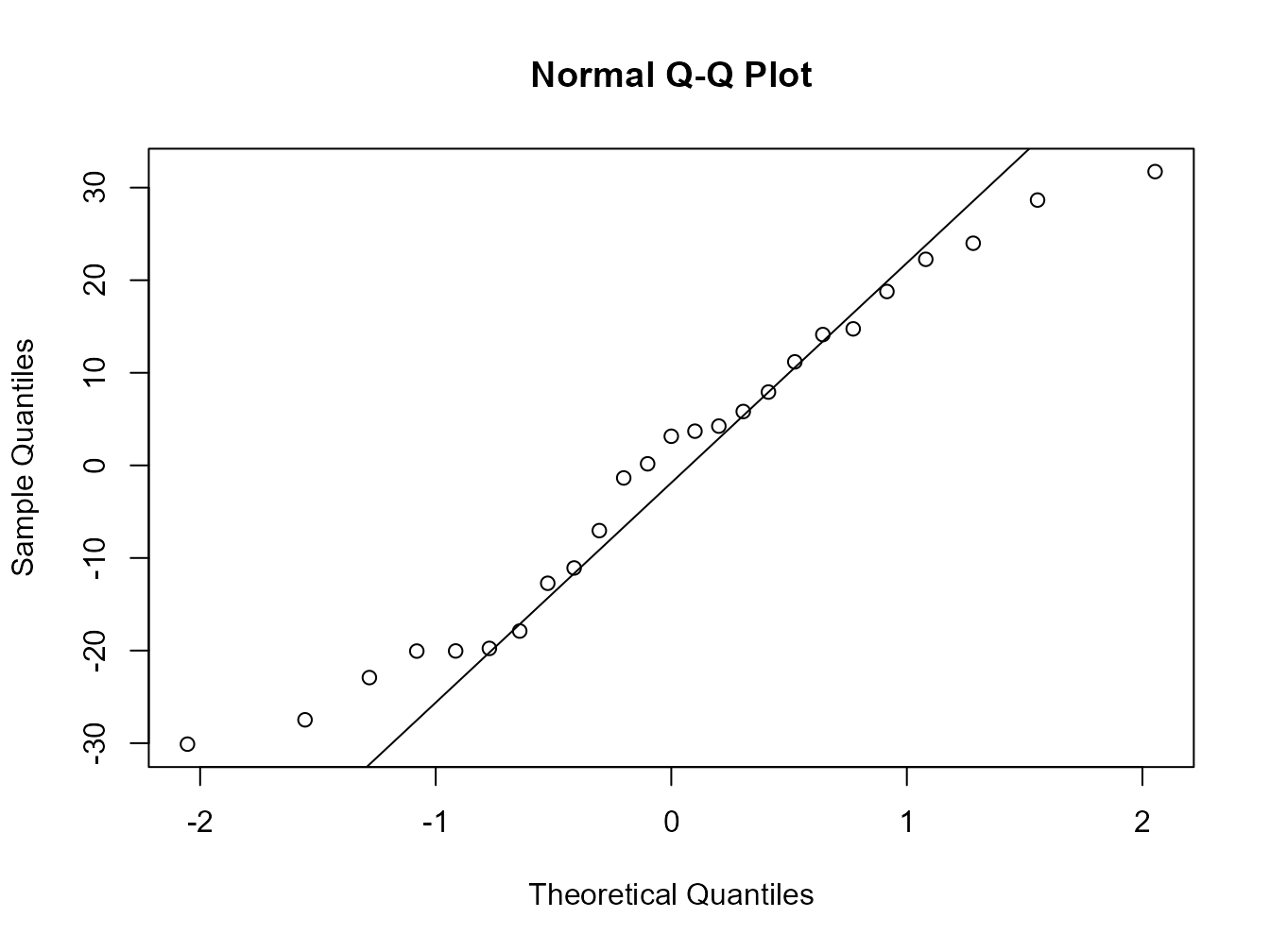
Alternatively, you can get R to do most of the hard work by using the plot() function on the model object smoke_lm. Before we do this we should tell R that we want to plot four graphs in the same plotting window in RStudio using the par(mfrow = c(2,2)). This command splits the plotting window into 2 rows and 2 columns.
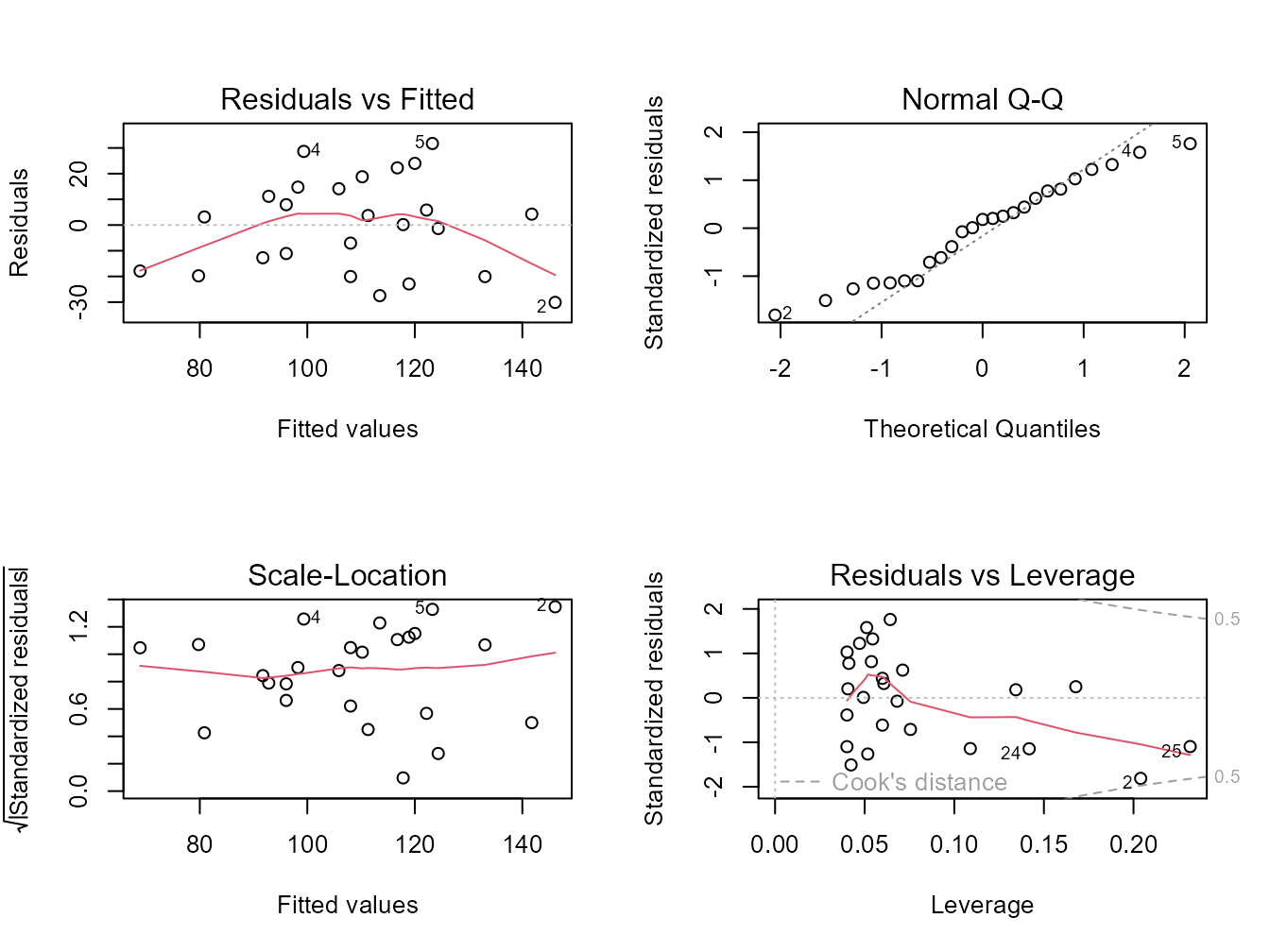
The first two graphs (top left and top right) are the same residual versus fitted and Q-Q plots we produced before. The third graph (bottom left) is the same as the first but plotted on a different scale (the absolute value of the square root of the standardised residuals) and again you are looking for no pattern or structure in the data points. The fourth graph (bottom right) gives you an indication whether any of your observations are having a large influence (Cook’s distance) on your regression coefficient estimates. Levearge identifies observations which have unusually large values in their explanatory variables.
You can also produce these diagnostic plots using ggplot by installing the package ggfortify and using the autoplot() function.
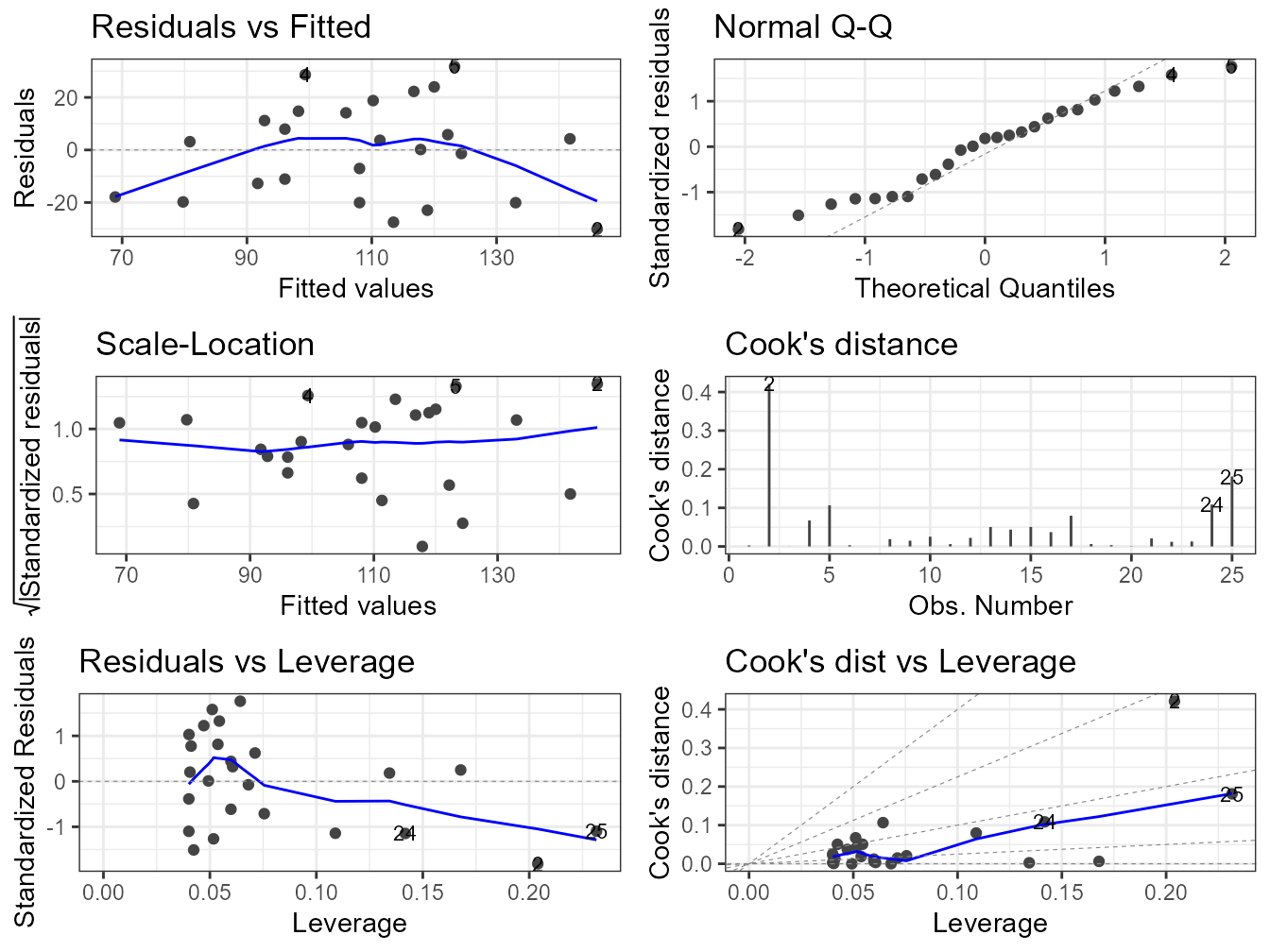
What you do about influential data points or data points with high leverage is up to you. If you would like to examine the effect of removing one of these points on the parameter estimates you can use the update() function. Let’s remove data point 2 (miners, mortality = 116 and smoking = 137) and store the results in a new object called smoke_lm2. Note, we do this to demonstrate the use of the update() function. You should think long and hard about removing any data point(s) and if you do you should always report this and justify your reasoning.
smoke_lm2 <- update(smoke_lm, subset = -2)
summary(smoke_lm2)
##
## Call:
## lm(formula = mortality ~ smoking, data = smoke, subset = -2)
##
## Residuals:
## Min 1Q Median 3Q Max
## -29.7425 -11.6920 -0.4745 13.6141 28.7587
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) -20.0755 23.5798 -0.851 0.404
## smoking 1.2693 0.2297 5.526 1.49e-05 ***
## ---
## Signif. codes:
## 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 17.62 on 22 degrees of freedom
## Multiple R-squared: 0.5813, Adjusted R-squared: 0.5622
## F-statistic: 30.54 on 1 and 22 DF, p-value: 1.488e-05There are numerous other functions which are useful for producing diagnostic plots. For example, rstandard() and rstudent() returns the standardised and studentised residuals. The function dffits() expresses how much an observation influences the associated fitted value and the function dfbetas() gives the change in the estimated parameters if an observation is excluded, relative to its standard error (intercept is the solid line and slope is the dashed line in the example below). The solid bold line in the same graph represents the Cook’s distance. Examples of how to use these functions are given below.
par(mfrow = c(2, 2))
plot(dffits(smoke_lm), type = "l")
plot(rstudent(smoke_lm))
matplot(dfbetas(smoke_lm), type = "l", col = "black")
lines(sqrt(cooks.distance(smoke_lm)), lwd = 2)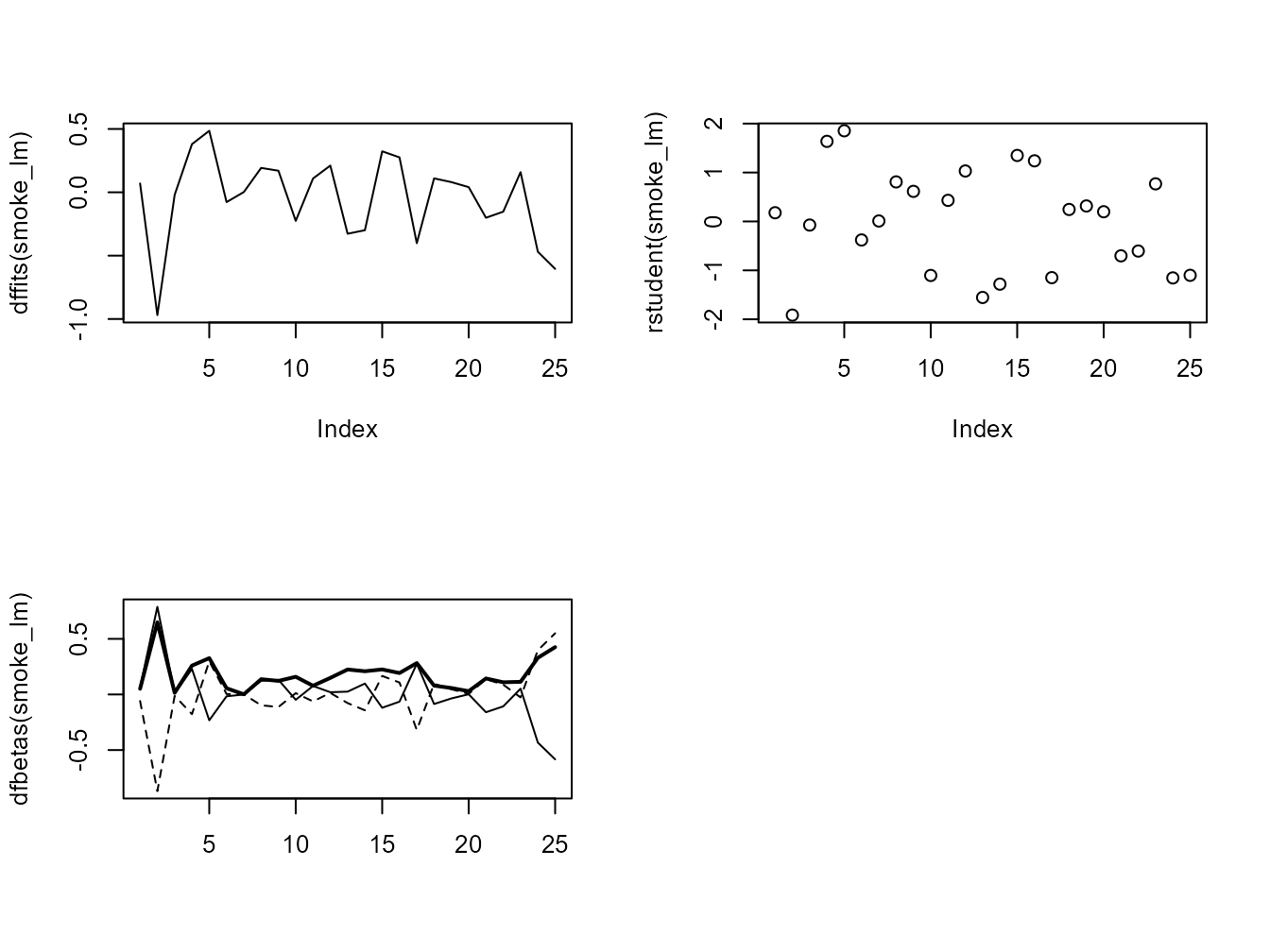
7.4 Other modelling approaches
As with most things R related, a complete description of the variety and flexibility of different statistical analyses you can perform is beyond the scope of this introductory guide. Further information can be found in any of the excellent documents referred to in the Additional Resources at the end of this guide and the cheat sheets.
A table of some of the more common statistical functions is given below to get you started.
| R function | Use |
|---|---|
glm() |
Fit a generalised linear model with a specific error structure specified using the family = argument (Poisson, binomial, gamma) |
gam() |
Fit a generalised additive model. The R package mgcv must be loaded |
lme() & nlme()
|
Fit linear and non-linear mixed effects models. The R package nlme must be loaded |
lmer() |
Fit linear and generalised linear and non-linear mixed effects models. |
The package lme4 must be installed and loaded |
|
gls() |
Fit generalised least squares models. The R package nlme must be loaded |
kruskal.test() |
Performs a Kruskal-Wallis rank sum test |
friedman.test() |
Performs a Friedman’s test |